京都府立医科大学
京都府立医科大学「多発性骨髄腫の病態解明に基く疾患制御戦略開発」プロジェクト
難治性造血器腫瘍である多発性骨髄腫(Multiple myeloma; MM)の病態形成や治療抵抗性には、極めて多彩で複雑な分子異常が複雑に絡み合っており、症例間での不均一性も高度であることが治療戦略開発を困難にする要因となっています。その打開を目指して、我々は
- 「細胞遺伝学的・分子生物学的に多様で不均一なMMにおける普遍的な異常の同定と制御戦略の開発」
- 「MMの多段階悪性化を促進する分子異常・分子制御異常の同定の制御戦略の開発」
- 「腫瘍免疫監視機構の破綻の克服」
などを中心に、多角的なアプローチでの挑戦を継続しています。
1.「細胞遺伝学的・分子生物学的に多様で不均一なMMにおける普遍的な異常の同定と克服戦略開発」プロジェクト
PDPK1/RSK2シグナル制御戦略開発プロジェクト
我々はMMにおいて非コードRNAであるmiR-375が病初期からほぼ普遍的にエピジェネティック制御異常によって発現低下状態にあること、そのため、PDPK1、IGF1、JAK2など重要な細胞シグナル分子の発現亢進をもたらすこと(Tatekawa S, Br J Haematol 2017)、さらにPDPK1の下流シグナル媒介分子であるRPS6KA3 (RSK2)が活性化する原因となることを見出しました。PDPK1/ RPS6KA3 (RSK2)シグナル経路の活性化は染色体異常や遺伝子異常のパターンに関わらずMMにおいて普遍的に多くの分子の制御に関わり、MMの病態形成を促進しています (Chinen Y, Cancer Res 2014; Shimura Y, Mol Cancer Ther 2012)。
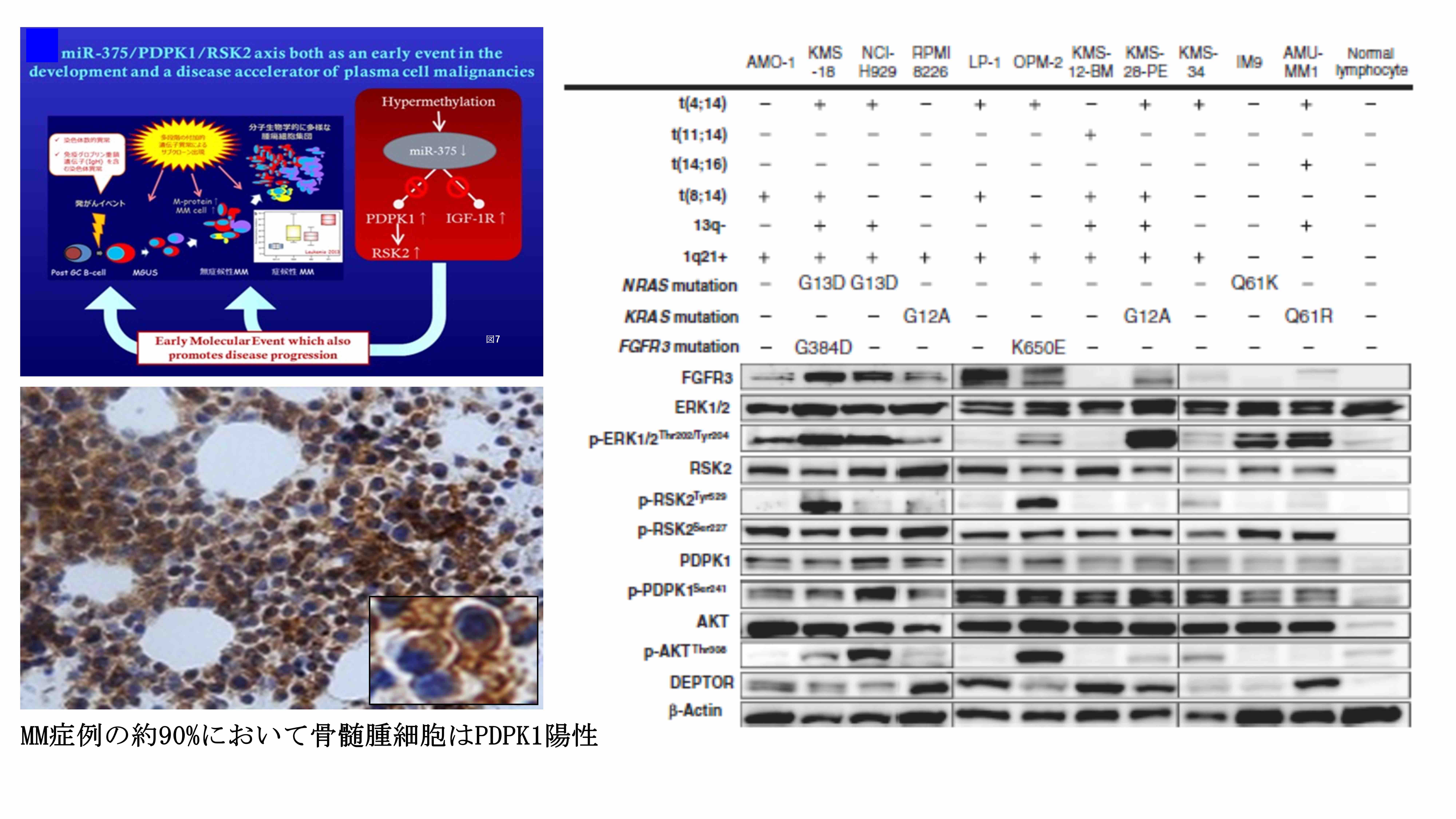
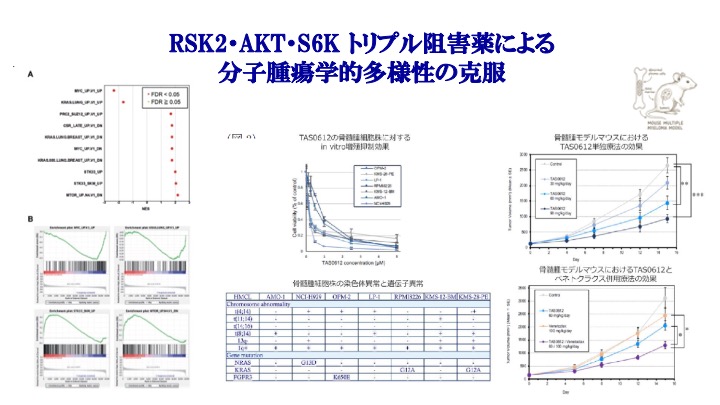
PDPK1はマスターレギュレーターとして多くのAGCキナーゼの活性を制御しています。われわれは、その被制御分子のうち、RSK2のN末端キナーゼ活性とAKTを制御することで、アポトーシス誘導分子であるBIMの誘導、BIDの活性化を伴うプログラム細胞死誘導を介した強力な抗腫瘍効果を骨髄腫細胞にもたらしうること、また、網羅的遺伝子発現解析、ならびにGene Set Enrichment解析などによる詳細な解析の結果、RSK2/AKT二重阻害は、それぞれの分子を単独で阻害した時と比し、有意に強力にMYC被制御遺伝子、mTOR被制御遺伝子、リボゾームバイオジェネシス関連分子、サイトカイン・ケモカイン誘導性細胞活性化分子など、骨髄腫細胞の増殖、生存、治療抵抗性獲得などを普遍的に支配する数多くの遺伝子・分子を多重に制御し、その結果、遺伝子異常や染色体異常のパターンの異なる多様な骨髄腫細胞に対して、普遍的に強力な抗腫瘍効果を誘導しうることを明らかにしました(Isa R, Int J Mol Sci 2022)。

こうした研究成果を理論的基盤とし、miR-375/PDPK1/RSK2シグナルによる造腫瘍性のメカニズムの更なる解析、ならびにそれらを標的とした創薬開発への展開をめざした創薬開発前臨床研究を産学連携研究として実施中であり、小分子化合物TAS0612の新規治療薬としての可能性を見出すとともに(Okamoto H, Leukemia 2025)、新規Protein degraderによるRSK2制御戦略の開発などに挑戦しています(Ide D, Yokoo H, Chem Pharm Bull 2026)。こうした経緯の中で、TAS0612が多発性骨髄腫の病態形成に極めて重要なMYC、RAS、mTORなどに支配される多くの遺伝子ならびに予後に関連する多くの遺伝子の発現を重複して多重に制御する分子薬理学的効果を有することを示しました。この効果はTAS0612による多発性骨髄腫の分子腫瘍学的複雑性や不均一性を凌駕し得る根拠として想定されます。また、TAS0612の応用法のひとつとして、急性骨髄性白血病や慢性リンパ性白血病に対して既に臨床実装されているBCL2阻害剤であるベネトクラクスとの併用効果を検討したところ、従前、ベネトクラクスは、多発性骨髄腫においては、t(11;14)染色体転座を有する約25%程度の症例のみ有効性を示せないことが既知でしたが、興味深いことにTAS0612との併用では、染色体異常の様式に関わらず強力な相乗的抗腫瘍効果を発揮しうることが判明しました。この結果をもとに、骨髄腫モデルマウスにおいてTAS0612単独ならびにベネトクラクスとの併用療法の効果を検討したところ(前臨床試験)、TAS0612は投与量依存的な抗腫瘍効果を発揮し、ベネトクラクスとの併用効果を発揮することが認められました。こうした結果は、今後の治療開発の新たなproof of conceptとしての可能性を示しています。
ガレクチン-9の抗骨髄腫効果
我々はβ-ガラクトシド結合性レクチンであるガレクチン9が細胞遺伝学的異常に関わらずMM細胞にJNKやp38MAPKなどのストレスキナーゼの活性化を介したアポトーシスを誘導すること、MM動物モデルにおいても抗腫瘍効果を発揮しうることを見出しました(Kobayashi T, Leukemia 2010)。この腫瘍効果発揮の過程でガレクチン9が結合するMM細胞上の標的は未解明であり、今後の研究課題です。
2.「多発性骨髄腫の多段階進展の分子メカニズムの解明と克服」プロジェクト
MMの疾患進行に関わる転写異常の研究:lncRNA PVT-1異常や、DCC遺伝子異常、CCND1遺伝子転写異常の発見
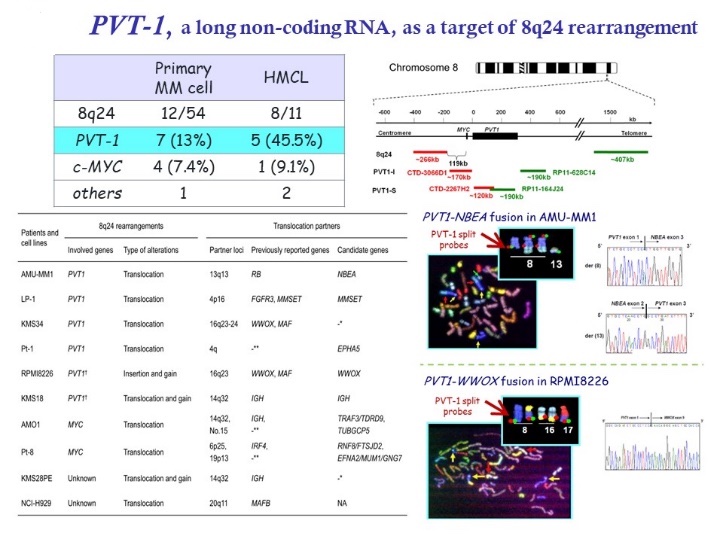
染色体8q24は二次的染色体異常の代表格の一つです。従来は、8q24転座では、がん遺伝子c-Mycの遺伝子再構成による過剰発現によって、疾患悪性度の高度化が誘導される機序が想定されていました。しかし、我々はMMでは多くの場合、c-Myc遺伝子の近傍に存在する長鎖非コードRNAであるPvt-1の切断が起こり、これがc-Mycの「過剰発現を誘導していること、多くの転座パートナー遺伝子の脱制御を誘発している可能性を見出しました(Nagoshi H, Cancer Res 2012)。
また、DCC遺伝子の転写制御異常がMGUSからMMへの病態進展に相関すること(Nagoshi H, Gene Chromosome Canc, 2015)、t(11;14)染色体転座に特異的なCyclin D1遺伝子の転写制御異常(Chinen Y, Exp Hematol 2020)、MMに特異的な分子異常・分子制御異常、EWSR1遺伝子発現亢進によるlet-7c, RA, AKT発現制御と病期進行の関係性(Nishiyama D, Int J Hematol 2020)などを見出し、これらの病態形成における機能的意義についての解明、診断法の開発を進めています。
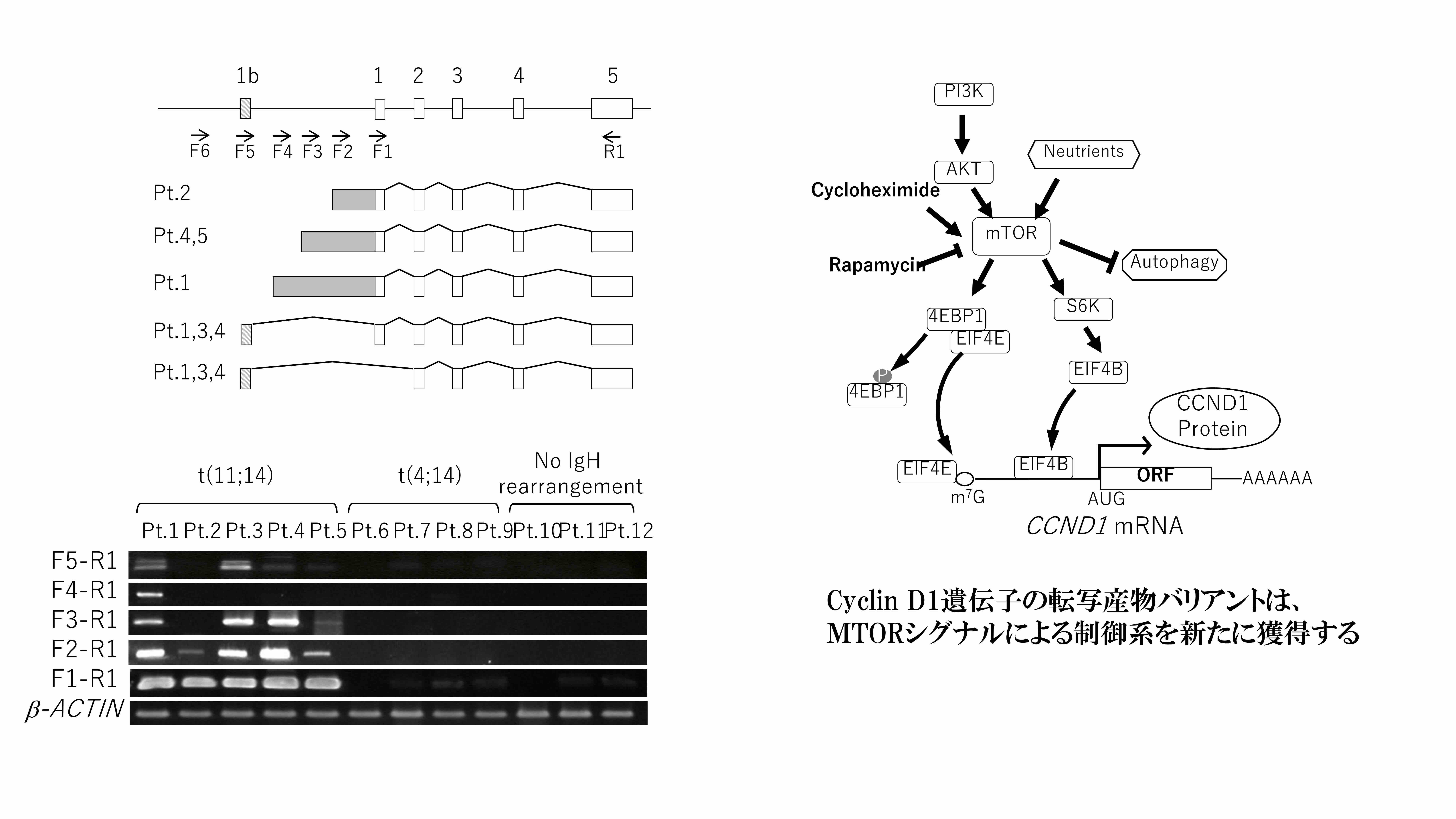
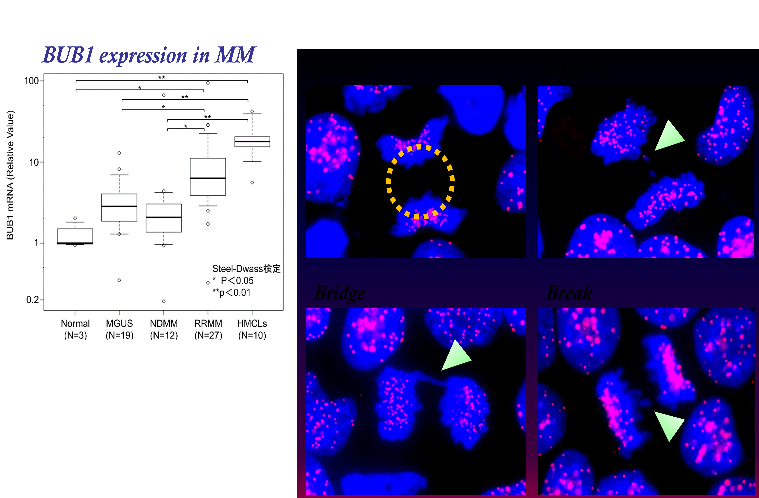
BUB1過剰発現と染色体分配制御異常による付加的染色体異常の獲得と病期進展の関係性を発見
他方、研究室ではMMの発症と進展に関わる染色体不安定性獲得によるクローン性進化のメカニズムの解明を目指した研究を行っています。そのなかで、MM細胞では細胞分裂期後期において高頻度に染色体分配異常が生じており、スピンドルチェックポイントを制御するチェックポイント分子の一つであるBUB1が病期進行とともに過剰発現し、正常の機能に反して、誤った染色体分配のままでの細胞分裂を許容するメカニズムが働いていることを見出しました(Fujibayashi Y, Cancers 2020)。今後、この誤作動を制御する戦略が開発できれば、がんの染色体クローン進展を阻止することが可能になるかも知れません。
3. MMにおける腫瘍免疫監視機構の破綻メカニズムの解明と克服戦略の開発
近年、各種の悪性腫瘍においてPD-1/PD-L1経路の治療標的化やCAR-T療法、BiTE療法、ペプチドワクチン療法など、腫瘍免疫監視機構の破綻の解除や、腫瘍免疫の活性化を介する新たな治療戦略の臨床実用化が競うように行われています。いずれも従来の治療では獲得できない有効性が期待できる一方で、治療奏効度や奏効期間はいまだに十分とは言えず、一方で治療に要する技術はしばしば高度であり、かつ、コストの負担も少なくなく、臨床現場のみならず医療経済の観点からも大きな課題となっています。
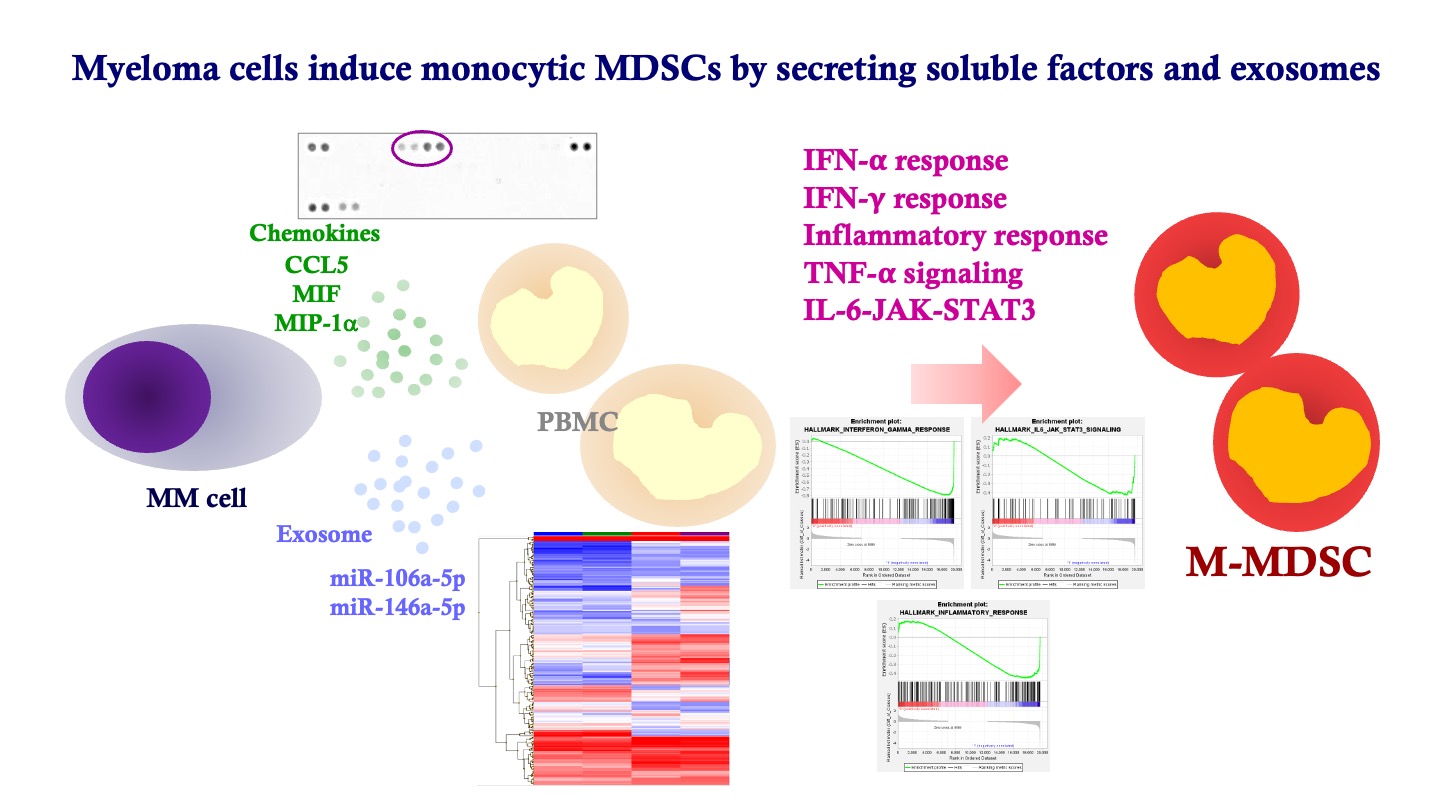
我々は、MMや悪性リンパ腫において腫瘍免疫監視機構の破綻を、より容易に解除しうる治療戦略の開発の実現を目指した研究を推進中です。なかでも、私たちが注目しているのは、様々な悪性腫瘍において増加し、制御性T細胞や制御性B細胞などの誘導を促進し、NK細胞や細胞障害性T細胞を抑制すると考えられている「骨髄由来抑制系細胞: Myeloid-derived suppressor cell (MDSC)」です。MDSCの存在は、一般的な免疫化学療法の効果のみならず、CAR-T療法などの細胞療法の効果を減弱させることが知られており、その制御戦略の開発は重要な研究課題です。
私たちは、MMなどの腫瘍環境において腫瘍細胞との共存状態において、正常末梢血単核球からMDSCが誘導されるin vitroシステムを構築し、疾患特異的なMDSC誘導の分子メカニズムを腫瘍細胞、末梢血単核球細胞のそれぞれについて網羅的に解析し、その制御戦略の開発研究を進めています。
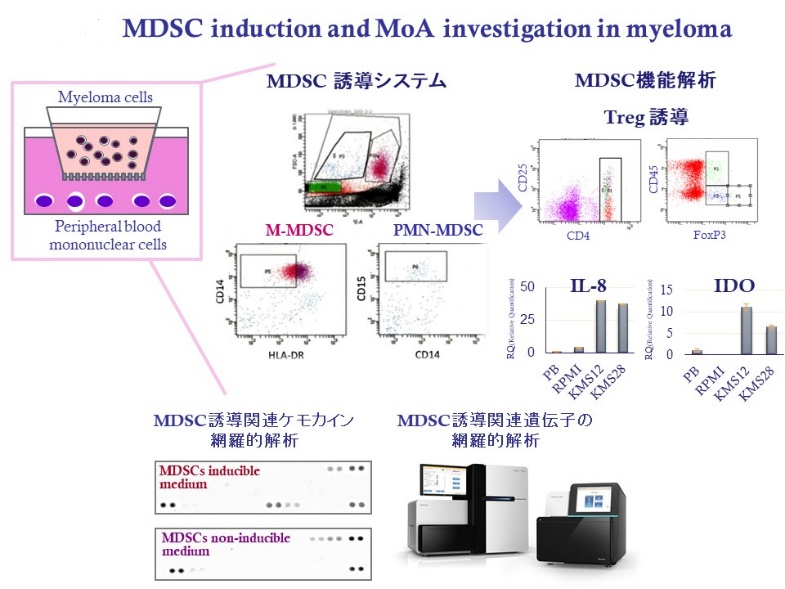
その過程において、MMでは骨髄腫細胞由来のCCL2とMIFが協調的に正常末梢血単核球からMDSC誘導し、疾患自ら免疫抑制環境の構築を促進していること、LenalidomideやPomalidomideなどの免疫調節薬は、骨髄腫細胞、末梢血単核球の双方に作用して、MDSC誘導を抑止しうることを見出しました(Kuwahara-Ota S, Br J Haematol 2020)。また、腫瘍由来エクソソームを介したCell-Cell Communicationによる免疫抑制環境構築の機序の存在、ならびに、その際にMDSC誘導に寄与するmiRNAを特定し、その分子生物学的機序として慢性インターフェロンシグナルによる免疫抑制性遺伝子群の発現による”Adaptive resistance”のメカニズムの一端を明らかにしました(Mizuhara K, Bj J Haematol 2023)。これらの包括的な解析を継続中であり、治療標的の同定やバイオマーカー確立、こうした免疫学的治療法の強化戦略の開発に向けた各種研究も同時に展開中です。